Percée en ingénierie des protéines : plus de 10M de données générées et une IA accélérée en seulement trois jours
Auteur: Mathieu Gagnon/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2026%2F04%2Fimage-2026-04-20T09-47-52-188Z.jpeg)
Le défi colossal de la complexité biologique

L’ingénierie des protéines se présente aujourd’hui comme un domaine de recherche idéal pour l’intelligence artificielle, comme le rapporte Rachel Leeson de l’Université Rice. Chaque protéine est constituée d’acides aminés. Pour optimiser sa fonction, les chercheurs doivent modifier cette structure originelle en remplaçant l’un des vingt acides aminés différents par un autre.
Si l’on prend l’exemple d’une protéine dont la longueur n’est que de cinquante acides aminés, cette opération conduit à environ 1,13×10^65 combinaisons potentielles à tester. Ce chiffre vertigineux correspond au nombre 113 suivi de 65 zéros, ce qui représente cinq fois plus de zéros qu’en compte un billion. Face à un tel volume de possibilités, les tests traditionnels en laboratoire deviennent matériellement impossibles.
Modéliser les combinaisons qui offriront les meilleurs résultats constitue un défi parfait pour la puissance de calcul massive de la technologie. Han Xiao, professeur de chimie, de biosciences et de bio-ingénierie à l’Université Rice, et directeur du SynthX Center, résume le problème technique : « L’un des plus grands goulots d’étranglement dans l’ingénierie des protéines guidée par l’IA n’est pas de concevoir des modèles d’apprentissage automatique. C’est de générer les données expérimentales correctes et en quantité suffisante pour les entraîner ». Le scientifique précise l’enjeu : « Pour l’ingénierie de l’activité protéique, qui optimise ce qu’une protéine fait, nous avions un problème très clair : il n’y avait tout simplement pas assez d’ensembles de données pour entraîner des modèles précis. »
L’émergence de la méthode Sequence Display
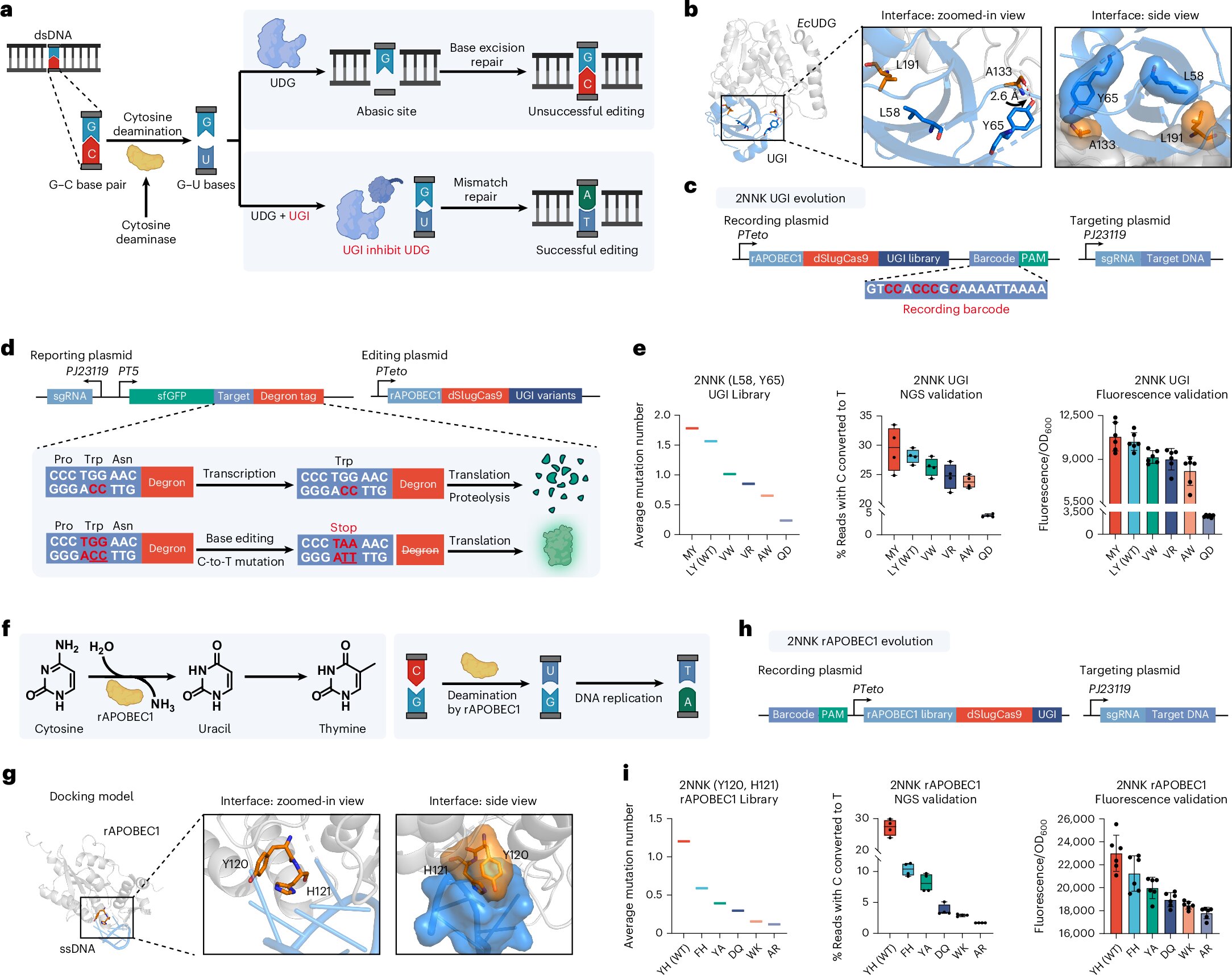
Afin de créer des algorithmes capables de prédire avec exactitude la manière d’optimiser l’activité d’une protéine, l’équipe du professeur Xiao a dû franchir un cap décisif. Il était impératif de produire au préalable un volume suffisant de données d’activité sur n’importe quelle protéine donnée pour nourrir le système. Cette prouesse technologique vient d’être documentée dans une récente publication de la revue Nature Biotechnology, datée de 2026.
En unissant leurs forces avec des collaborateurs de l’Université Johns Hopkins et de Microsoft, les chercheurs ont partagé une approche inédite qui a fourni les données requises et permis de créer des modèles précis en seulement trois jours. Cette technique novatrice, baptisée « Sequence Display », s’avère d’une efficacité redoutable puisqu’elle peut générer plus de dix millions de points de données lors d’une seule expérience.
Ces masses d’informations sont ensuite injectées dans des modèles d’intelligence artificielle de langage protéique. Linqi Cheng, étudiant diplômé de l’Université Rice et premier auteur de l’étude, explique la démarche : « Nous avons pu développer un système de code-barres basé sur l’activité qui enregistre l’activité des variantes protéiques individuelles et génère le type d’ensemble de données nécessaire pour entraîner un modèle d’apprentissage automatique ». Le jeune chercheur observe le résultat direct : « Ensuite, le modèle a été capable de prédire des mutations qui ont considérablement amélioré l’activité de la protéine que nous étudiions. »
La preuve de concept par la manipulation de l’ADN

Pour valider la viabilité de leur système, les chercheurs ont jeté leur dévolu sur une petite protéine CRISPR-Cas. Cette molécule spécifique était particulièrement appréciée pour sa taille réduite, bien qu’elle présentât des limites notables dans son activité lorsqu’il s’agissait de cibler des segments d’ADN à couper. L’ambition de l’équipe consistait à identifier une version capable de sectionner une plus grande variété de cibles génétiques.
Le protocole expérimental a débuté par la mutation de l’ADN qui code pour la protéine Cas9, générant ainsi de multiples variations. Un code-barres d’ADN vierge a été méticuleusement attaché à chaque variante, accompagné d’un éditeur spécial conçu pour modifier ce marqueur en réaction directe au niveau d’activité de la protéine. La mécanique est implacable : à mesure que les niveaux d’activité de la protéine augmentaient, ceux de l’éditeur suivaient la même trajectoire.
Par conséquent, les variations protéiques les plus actives enregistraient les changements les plus importants au niveau de leurs codes-barres. Une fois cette étape biochimique achevée, les codes-barres d’ADN ont été analysés par un séquençage de nouvelle génération. Cet appareillage scanne le code-barres et classe rigoureusement chaque séquence en fonction de son niveau d’activité, fournissant une cartographie précise de l’expérience.
Une synergie indispensable entre machine et laboratoire

Cette approche méthodologique souligne une dépendance fondamentale entre la biologie physique et la puissance algorithmique. Linqi Cheng insiste sur cette interconnexion cruciale : « L’IA ne remplace pas l’expérience ici. Elle dépend au contraire de l’expérience ». Le doctorant détaille la répartition des rôles : « Sequence Display nous donne la fondation de données, et les modèles nous aident à chercher de solides candidats dans un espace de données beaucoup plus vaste. »
Loin de s’arrêter à la seule molécule CRISPR-Cas, l’équipe a déployé ce même protocole sur un panel plus large. Le processus a été répété avec succès sur d’autres protéines, incluant les aminoacyl-ARNt synthétases, la cytosine désaminase ou encore l’inhibiteur de l’uracile glycosylase.
Pour chacune de ces tentatives complexes, l’expérience de marquage par code-barres a rempli son objectif initial de manière systématique. Elle a généré à chaque fois un nombre de points de données amplement suffisant pour entraîner efficacement les modèles d’intelligence artificielle, confirmant la reproductibilité de la méthode.
Vers une nouvelle ère de thérapies de pointe

L’impact de ces recherches dépasse le simple cadre de l’optimisation moléculaire en laboratoire. Han Xiao, qui officie également en tant que boursier du Cancer Prevention and Research Institute, met en perspective la portée de ces travaux fondamentaux : « Ce que cette approche fournit est un cadre pratique pour intégrer l’IA à l’ingénierie des protéines ».
Le directeur de recherche envisage une évolution profonde des méthodes de travail : « Plutôt que de s’appuyer sur l’apprentissage automatique comme une solution autonome, nous le couplons avec une plateforme expérimentale qui génère des données d’entraînement de haute qualité. Cette synergie permet une découverte plus efficace d’outils de recherche avancés et de protéines thérapeutiques de nouvelle génération. »
L’ensemble de ces avancées majeures a été consigné avec rigueur sous le titre de publication « Sequence Display enables large-scale sequence–activity datasets for rapid protein evolution ». Ces résultats signés par Linqi Cheng et ses co-auteurs sont documentés dans l’édition 2026 de Nature Biotechnology, identifiables par le numéro DOI 10.1038/s41587-026-03087-3.
According to the source: phys.org
/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2026%2F04%2Fimage-2026-04-12T10-30-32-709Z.jpeg)
/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2026%2F04%2Fimage-2026-04-12T10-23-45-794Z.jpeg)
/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2026%2F03%2Fimage-2026-03-19T14-07-02-384Z.jpeg)
/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2026%2F03%2Fimage-2026-03-14T12-30-15-248Z.jpeg)
/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2025%2F11%2Fimage-1763110572171.jpg)
/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2025%2F11%2Fimage-1762938670629.jpg)
/https%3A%2F%2Flanature.ca%2Fapp%2Fuploads%2F2025%2F09%2F2048px-Australopithecus_sediba_Fundort_Malapa.jpg)